
Nos dernières actualités

Silence radio aborde de manière ludique la santé mentale auprès des 15-25 ans. À l’occasion de la Semaine nationale de...

La médecine se veut universelle. Pourtant, dans les faits, une réalité persiste : les femmes restent encore...

245 847 : c’est le nombre de médecins en activité inscrits au tableau de l’Ordre des médecins au 1er janvier 2026.

La Sécurité sociale accuse un déficit de 21,6 milliards d’euros en 2025, dont 15,9 milliards pour la branche maladie....

Comment favoriser le maintien en autonomie des personnes âgées et en situation de handicap ? Au total, 80...
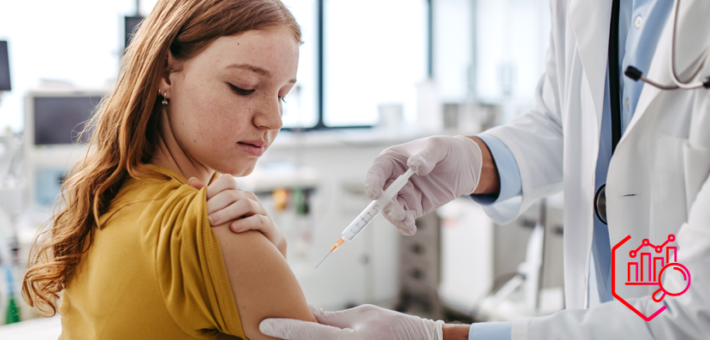
61,6%, c’est la part des jeunes filles de 15 ans ayant reçu une première dose de vaccin contre le papillomavirus...

Le centre mutualiste de l'obésité d'Aésio Santé propose un parcours de soins structuré et pluridisciplinaire, comprenant...

C’est un titre qui résonne comme une locution familière et rassurante. “Et la santé, ça va ?” est le nom du premier...

Parmi les thématiques abordées dans les ateliers territoriaux, dans le cadre de la phase de co-construction des Etats...

La consultation des États généraux de la santé et de la protection sociale a mis en lumière une attente forte des...

Mercredi 15 avril 2026 s’est tenu un nouvel atelier territorial à Montpellier, organisé par l’Union régionale de la...

20,2 millions : c’est le nombre d’hospitalisations réalisées en 2024 dans les établissements de médecine, chirurgie et...

Un atelier des États généraux de la santé et de la protection sociale s’est tenu le 31 mars, à Paris, lors du Congrès de...

De la grossesse aux deux ans de l’enfant, les 1 000 premiers jours constituent une période décisive. Sans que tout s’y...

96 % : c’est le taux de satisfaction global des patients des centres de santé mutualistes en 2025, selon...